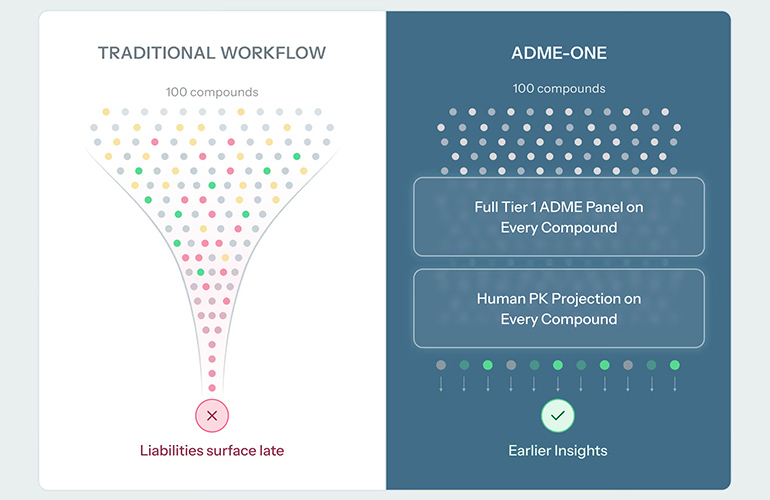
For years, many small-molecule teams have treated comprehensive Absorption, Distribution, Metabolism and Excretion (ADME) profiling as a lead-optimization step. That is, it serves as a place to spend heavily once a lead series has already emerged. Now, Ginkgo Datapoints, Tangible Scientific and Inductive Bio are betting that AI, automation and tighter compound logistics can move that decision point much earlier.
The new ADME-One platform packages Ginkgo’s automated Tier 1 assays, Tangible’s compound-management workflow and Inductive Bio’s human pharmacokinetic projection into a single service aimed at hit identification. The aim is to give medicinal chemists an earlier read on potency, ADME, projected human PK and even dose, before months of synthesis cycles narrow a program around the wrong compounds.

Alex Taylor, Ph.D.
The guiding question behind the platform was whether AI and automation could make the whole assay setup substantially more cost-effective. “We asked: Could we pull together all the assays needed to get your first projection of human PK at a price point where you’d now be doing this on most, if not all, of the compounds coming through?” said Alex Taylor, Ph.D., head of medicinal chemistry at Inductive Bio. The idea, he said, was to get that capability into the first tier of assays a program runs: “Can you get your potency plus the ADME you need, and put those together to get your initial estimate of human PK, and even dose?”
Ginkgo Datapoints runs all five Tier 1 assays (microsomal stability, cell permeability, kinetic solubility, CYP inhibition and plasma protein binding) end to end in its automated lab in Boston. Inductive Bio’s Compass platform rolls those individual readouts into a single human PK projection that teams use to rank compounds, while Tangible Scientific takes custody of the physical samples, handling intake, plating and real-time tracking for each order. The partners “did a lot of validation behind the scenes to make sure we’re still getting really high-quality data,” Taylor said.
Earlier ADME and PK context
One growing direction in the med chem field is to focus on human dose as early as possible. “Experienced medicinal chemists will tell you up front that dose is ultimately the thing you want to optimize for,” Taylor noted.
For instance, a Hepatology analysis led by FDA-affiliated researchers found that high daily dose, especially combined with high lipophilicity, was associated with significantly elevated risk of drug-induced liver injury, the so-called rule-of-two; the same work cautions that many high-dose drugs are nonetheless safe, so the relationship is a risk signal rather than a verdict. A separate registry study found drugs dosed at 50 mg per day or more carried higher rates of liver failure, transplant and death than those below 10 mg. Lower doses also tend to be easier to formulate and to take, and simpler dosing regimens are associated with better patient adherence. “But it’s historically been hard to get all the data together to understand what your dose is,” Taylor said.
In addition to the data integration challenge, drug discovery routinely turns up potent compounds that score poorly on metabolic stability.
Here, triazole antifungals serve as an example. Fluconazole is small and polar, binds plasma protein only weakly, and clears largely unchanged through the kidneys, while itraconazole sits at the opposite extreme. It is highly lipophilic, more than 99% protein-bound, extensively distributed into tissue, and cleared by hepatic metabolism. On an assay-by-assay scorecard the two profiles look irreconcilable, yet both became established, widely used oral antifungals.
Speaking broadly, Taylor noted that “sometimes compounds you think aren’t good enough to go forward, because they don’t meet your criteria for potency or metabolic stability, actually have a balance of all the properties such that they could go forward.”
The cost-and-speed driver
Cost discipline shapes how most discovery teams operate, and the pressure has only ramped up. “The mantra in the whole tech and pharma world right now is to stay lean, be cost-competitive, be cost-conscious,” Taylor said. That pressure has long justified the screening-funnel approach, and it is also what keeps teams from gathering the broader data set that would clarify human PK and dose.
One factor that helped change the economics was the automation underneath the assays. “The ability to run many compounds, a whole plate full, using the robotics on a weekly basis and turn that around in an automated way, that’s what’s made the difference,” he said.
The ADME-One platform’s pricing sits below industry standard and is pitched to beat offshore CROs, a response to U.S. and European developers moving preclinical work back onshore amid the BIOSECURE Act and rising demand for data sovereignty. The workflow runs entirely in the U.S. and returns results in days rather than weeks.
The consortium model and data security
Beyond where the work runs, the consortium raises a separate question, and it lands on Inductive, the partner behind the PK projections: how do you improve the shared ML models on customers’ chemistry without exposing any single customer’s compounds? Taylor’s answer described Inductive’s own data architecture, a consortium that Inductive runs separately from the three-company ADME-One partnership. “The founders [of Inductive] put together a consortium model, a legal framework where all our partners can pool their data securely, and no partner can see anyone else’s data,” Taylor said. That pooled data trains Inductive’s global models. When a customer contributes its own results, the company fine-tunes a local model on top of the global one, which Taylor said usually delivers a strong performance gain.
Taylor said the team put substantial engineering into making the pooled data impossible to reverse-engineer: “you can’t look at what’s similar to your compounds as a way of backing out what’s in the consortium.”
Toward a virtuous cycle
The payoff, in his description, is a flywheel in which more participating data broadens the chemical matter behind the global models. “The flywheel works by continually feeding the consortium with more and more breadth of chemical matter, which helps everybody,” he said.
What fills that consortium is the day-to-day loop inside each program, which Taylor described as a “virtuous cycle.” Chemists design molecules on the Inductive platform while seeing predicted ADME parameters and a visualization of how those predictions roll up into a human PK curve. Selected compounds then move through synthesis and into the Inductive, Tangible and Ginkgo ADME-One platform, where the experimental results show how well the ADME and human PK predictions held up.
“That helps train the models, makes them better the next time people are designing compounds, and it improves from there,” Taylor said.
Still, Taylor framed AI’s role in drug discovery as a prioritization tool grounded in experimental work. “Drug discovery is science at the end of the day, and science is not engineering,” he said. “There’s still so much that needs to happen empirically. You can give your best guess of what a compound is going to do, but at the end of the day you need to reduce it to practice, synthesize it, and test it.”
That makes prediction useful in a narrower, practical sense: deciding which expensive, slow compounds deserve to be made first. “The ability to predict any of the parameters going into those decisions is incredibly useful,” Taylor said, “because no matter what the compound is, it always takes longer than you want to make, and it’s always expensive, and teams have a limited budget.”
Filed Under: Drug Discovery