 A clinical trial of the most common sickle cell anemia drug, hydroxyurea, was halted a year early this winter because of “overwhelming evidence of benefit,” reported University of Nebraska Medical Center pediatric researcher Stephen Obaro, M.D., Ph.D., in The Lancet.
A clinical trial of the most common sickle cell anemia drug, hydroxyurea, was halted a year early this winter because of “overwhelming evidence of benefit,” reported University of Nebraska Medical Center pediatric researcher Stephen Obaro, M.D., Ph.D., in The Lancet.
Hydroxyurea is “the only readily available disease-modifying therapy for patients with sickle cell,” Principal Investigator Russell Ware, M.D., Ph.D., told Drug Discovery & Development, so this was good news to patients and clinicians. Ware is director of hematology at the Cincinnati Children’s Hospital.
“Recently published National Institutes of Health (NIH) evidence-based guidelines [also] suggest it should be used more often,” Ware added.
But hydroxyurea does not solve all problems. It ultimately fails 50 percent of patients (if this number may drop with more widespread and knowledgeable use of the drug.) Average sickle cell patients only live into their 40s (if this number, too, could change with more widespread and knowledgeable use of the drug.) And while “[the drug] is widely available in the U.S. and Europe, it is not in low-resource countries,” Ware told Drug Discovery & Development. Given that two-thirds of the 305,800 newborns with sickle cell anemia live in Africa, this is one of many problems.
So other approaches are being investigated globally, including a pioneering genetically engineered stem cell clinical trial that recently enrolled its first patient.
Hydroxyurea
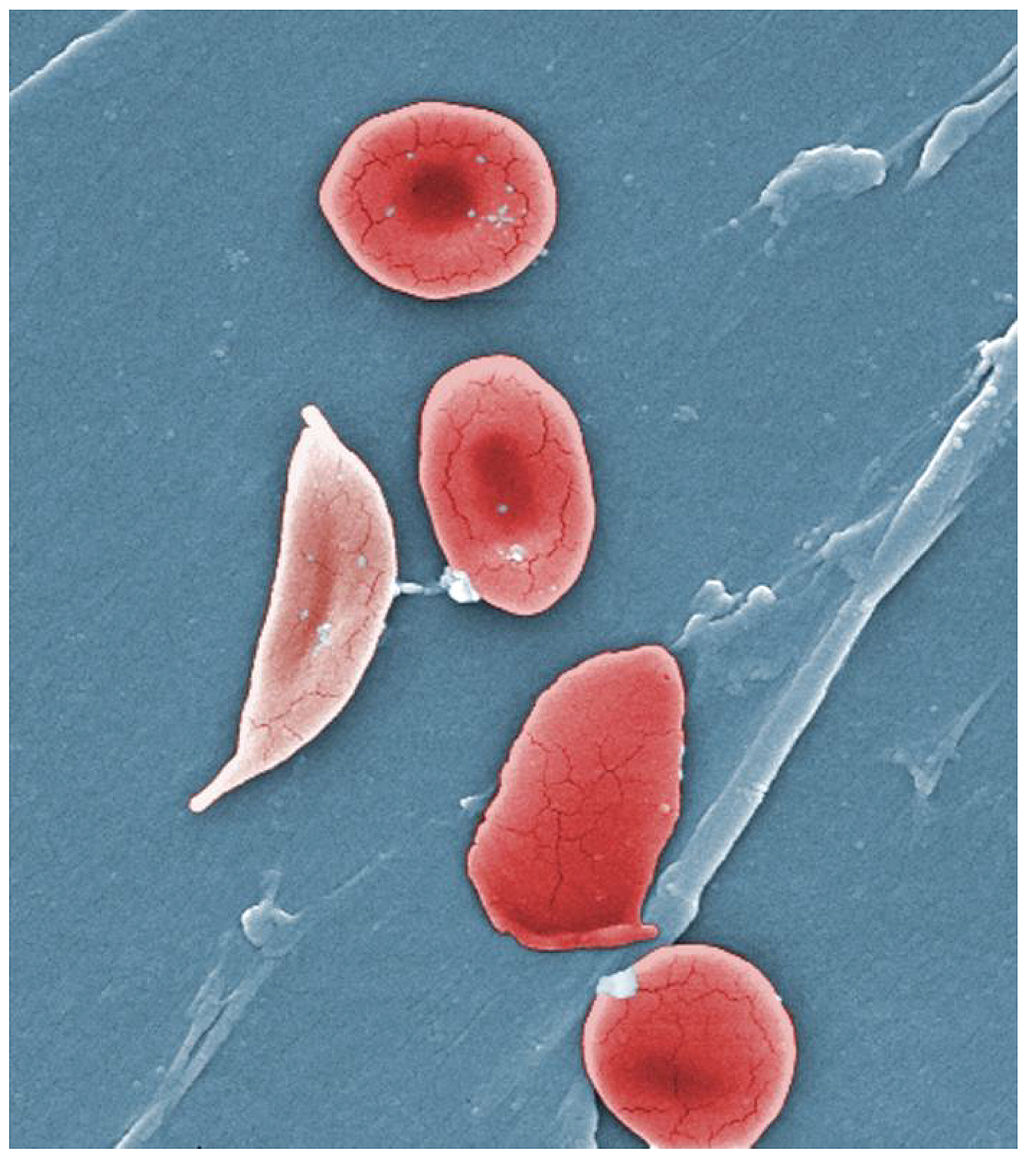
Sickle cell anemia occurs as the result of an inherited mutation in the oxygen-carrying hemoglobin (Hbs) gene. This causes red blood cells to become sticky, and form a sickle shape, which impairs their ability to carry oxygen, and causes them to aggregate and form constant blockages in the blood stream. As red blood cells become sickled and die, patients are chronically anemic. Blood vessel blockages called vaso-occlusions can cause pain in the chest, arms, and legs—and strokes. By age 20, about 15 percent of children suffer strokes, and half have nervous system disorders leading to serious cognitive dysfunction.
Establish your company as a technology leader. For 50 years, the R&D 100 Awards, widely recognized as the “Oscars of Invention,” have showcased products of technological significance. Learn more.
Blood transfusions can prevent strokes by flooding patients’ bodies with normal red blood cells. And hydroxyurea, an anti-metabolite, has been found in recent years to increase hemoglobin levels and reduce pain when taken daily. It does this by prompting the body to make another hemoglobin, fetal hemoglobin, and to reduce white blood cells, which thins the blood and makes it less prone to clotting.
Still, as noted, there are problems. In the halted trial, it was quickly found hydroxyurea prevents strokes as well as blood transfusions, a key development. Still, the reduced  white-blood cell counts can leave some patients open to infections. And—again—the drug doesn’t work on all patients.
white-blood cell counts can leave some patients open to infections. And—again—the drug doesn’t work on all patients.
So research has continued. “There are several new research trials designed to prevent or shorten acute vaso-occlusive events, which typically present as pain and acute chest syndrome,” Ware told Drug Discovery & Development. Indeed, of 436 sickle cell clinical trials registered at the NIH, 60 are devoted to vaso-occlusive events.
The only sure cure comes from stem cell transplants, which involve removing blood stem cells from patients’ bone marrow, and replacing them with blood stem cells from healthy matched donors. This way, patients receive new blood and immune systems.
First genetically engineered stem cell trial for sickle cell
But this isn’t perfect either. Transplants of stem cells from donors cost hundreds of thousands of dollars, Ware noted. And only 15% of sickle cell patients have matched siblings. Unmatched cells can be rejected by the immune system. So “there are ongoing efforts to make curative therapy via stem cell transplantation more safe and available to more patients,” Ware told Drug Discovery & Development.
At the head of the pack are University of California, Los Angeles (UCLA) immunologist Donald Kohn, M.D., and UCLA hematologist Gary Schiller, M.D., who recently launched the first U.S. genetically engineered stem cell trial for sickle cell. In this approach, as they outlined pre-clinically in a 2013 Journal of Clinical Investigation, their team removes a few stem cells from patients’ bone marrow, and uses genetic engineering to add a healthy version of the defective hBB gene to them.
Specifically, they use a defanged lentiviral vector to ferry the healthy new genes into patients’ stem cells. Then they give the cells back.
In mice, the human cells worked for a few months, then were rejected by the non-human immune systems. But researchers expect the gene to last much longer in patients, since they are receiving their own cells back. No rejection should be involved. Only a small percent of cells take up the healthy gene, but this should not matter. What is needed is a steady continuous supply of healthy cells to counteract some of the deadly effects of sickle cell.
Since blood stem cells are permanent residents of the bone marrow, that steady continuous supply should be achieved.
“Neat and exciting”
Schiller warned Drug Discovery & Development that it is “pretty early. One patient is entered, and others are being evaluated.”
But all told, he said: “It is a neat and exciting trial.”
Kohn has apparently seen success with two gene therapy clinical trials treating another genetic disorder: ADA-SCID, or adenosine deaminase-deficient severe combined immune deficiency. The results have not yet appeared in a peer-reviewed journal. But UCLA says 18 patients have been successfully treated for the disorder. In the trials, a healthy gene was added to some stem cells, as will occur in the sickle cell trial.
But in the ADA trials, clinicians could administer drugs to elicit the stem cells from the bone marrow. As sickle cell patients go into sickle cell crisis on those drugs, Schiller’s crew will have to remove the cells from the bone marrow, a trickier affair.
New approaches on horizon
Yet already, improvements are brewing there, too. Kohn this month published a paper in Blood reporting that he has fixed the mutation causing the disease, using a genetic engineering tool called zinc finger nucleases. This may mean that in the future, he won’t have to add an extra copy of the gene to cells—he will be able to simply fix them.
Also this month, immunologist Linzhau Cheng of Johns Hopkins University published a paper in Stem Cells reporting his team also corrected the sickle cell mutation in blood stem cells. His approach used the CRISPR gene-editing technique, and the cells were derived from iPSCs (induced pluripotent stem cells).
For a list of all sickle cell clinical trials registered with the NIH, go to clinicaltrials.gov.
Filed Under: Drug Discovery


