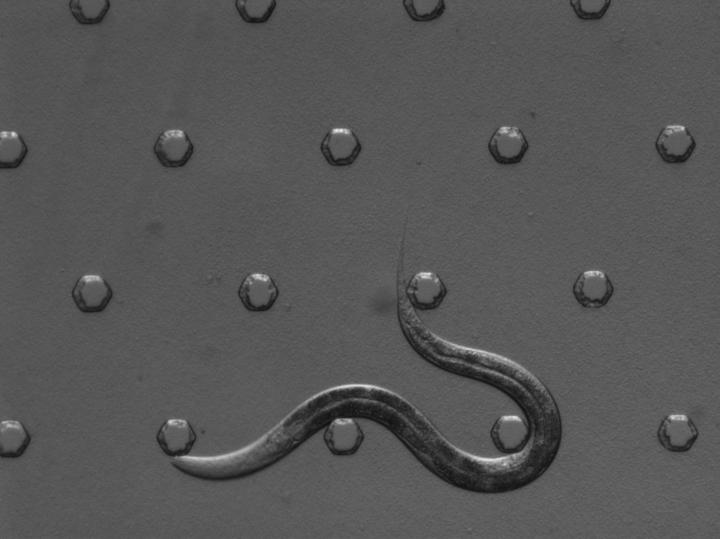
Using selectively engineered C. elegans — transparent worms about as long as a pinhead — Anne Hart, a professor of neuroscience at Brown University, discovered that two different kinds of motor neurons that die in people with ALS may die in different ways.
Brown University researchers have uncovered new clues about the progression of Amyotrophic Lateral Sclerosis (ALS), a surprisingly common disease that causes the death of motor neurons that control voluntary muscles such as those involved in walking, talking, chewing or breathing.
A team led by Anne Hart, a professor of neuroscience at Brown, discovered that two different kinds of motor neurons that die in patients may die in different ways — an important insight for understanding the disease and, eventually, finding a cure. Their work was published on Monday, Oct. 8, in the journal PLOS Genetics.
ALS is characterized by the degeneration of motor neurons in the spine that directly control muscle movement. But in some people with ALS, neurons in the brain that issue commands to these spinal motor neurons also die. It’s not clear why both types of neurons are affected in some people with ALS, but not others.
“Our results raise the possibility that the glutamatergic neurons in the brains of some ALS patients die in ways that are somehow different than how the spinal cord neurons die,” said Hart, a researcher at Brown’s Carney Institute for Brain Science. “Before, we all assumed that both kinds of neurons died the exact same way.”
And the implications could be significant, Hart said — this is the first clue that future treatments developed for spinal cord neurons might not cure all people with ALS, because they won’t help affected neurons in the brain.
Though many cases of ALS don’t have a clear genetic component, about 1 percent of people with ALS have mutations in SOD1, a protein involved in breaking down naturally occurring free radicals from oxidative stress. Hart’s research team precisely and selectively engineered C. elegans– transparent worms about as long as a pinhead — so that the worm’s SOD1 gene would contain a mutation such as those found in these people with ALS.
The results could explain why only spinal neurons are affected in some people, yet neurons in both the spine and the brain die in others.
More research needs to be done to see if the findings from worms will hold true in mammalian brains and lead to a better understanding of why neurons degenerate in people with ALS.
“We certainly can’t prove this in worms,” Hart said, “but it opens up a whole new way of looking at ALS.”
Specific neuron death
The work in Hart’s lab to develop a new ALS model has been a long-term effort. Seven years ago, postdoctoral researcher Jill Yersak began engineering different ALS mutations into worms. Worms have neurons very similar to human neurons and are less expensive and produce results faster than mice or other mammals, Hart said.
The project was completed by graduate student Saba Baskoylu. Then, the Brown research team ran numerous tests to see how the different patient versions of the SOD1 protein affected neuron function, motor neuron death and worm behavior.
Hart’s team found that four patient gene mutations caused neurodegeneration after oxidative stressin a type of neuron similar to those in the human spine, likely through increased toxic protein accumulation that doesn’t happen with the normal protein. However, two patient gene mutations also caused degeneration in a different type of neuron –similar to the neurons in the human brain–in part because the mutant protein no longer functioned correctly during oxidative stress.
The other kinds of neurons were healthy in the new ALS models, even after oxidative stress, which is very much like the specificity of neuron death in people with ALS, said Hart. In contrast, previous worm models weren’t very specific — almost any worm neuron could be killed by the patient version of SOD1.
Precise worm models
These earlier worm models were made by adding extra copies of a patient disease gene to the worms, which would then express the patient version of SOD1 at high levels. Now, thanks to new genetic tools like CRISPR/Cas9, directly editing the genes of worms and other animals is affordable and reliable. That allows scientists to make more precise models compared to simply adding extra copies of a gene, said Hart.
Hart’s goal was a more precise disease model that would let her group study the earliest events in ALS, by using these tools to change one “letter” in the worms’ standard “blueprint” for the SOD1 protein. The worms should have normal amounts of the protein and no extra gene copies.
The team accomplished their goal and discovered that the glutamatergic neurons –similar to the ALS-affected neurons in humanbrains — and the cholinergic neurons –similar to the spinal neurons — in worms degrade for different reasons. They will do more research on these worm models.
“We can now use these new ALS models to find other proteins and genes that we can use to stop neurodegeneration in worms,” Hart said. She plans to use the models to test many different small molecules for potential therapeutic drugs and to find other genes whose inactivation will suppress neurodegeneration. Then, collaborators, including those in other Brown labs, can test these genes in mice or human cell cultures, with the hope of helping people with ALS.
“ALS is complicated, you can see why it’s taking everyone a while to figure out what’s going on,” Hart said.
Filed Under: Neurological Disease