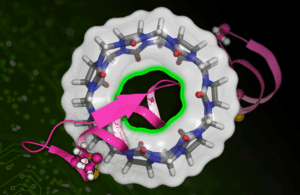
MIT researchers have developed a machine learning-based technique to more quickly calculate the binding affinity of a drug molecule (represented in pink) with a target protein (the circular structure). [Image courtesy of MIT News]
MIT researchers are touting a new machine-learning technique called DeepBAR that can quickly calculate the binding affinities between drug candidates and their targets.
DeepBAR produces precise calculations in a fraction of the time compared to conventional techniques, according to the researchers. They think the software could potentially accelerate drug discovery and protein engineering.
“Our method is orders of magnitude faster than before, meaning we can have drug discovery that is both efficient and reliable,” Bin Zhang, an MIT chemistry professor and a co-author of a new paper describing the technique, said in an MIT news release.
The research, which NIH partially funded, appeared today in the Journal of Physical Chemistry Letters. The study’s lead author is Xinqiang Ding, a postdoc in MIT’s Department of Chemistry.
Drug discovery researchers measure the affinity between a drug molecule and a target protein by a quantity called the binding free energy: The smaller the number, the stickier the bind. “A lower binding free energy means the drug can better compete against other molecules, meaning it can more effectively disrupt the protein’s normal function,” Zhang said.
A drug candidate’s binding free energy helps indicate a drug’s potential effectiveness, but it’s a difficult number to figure out. One computing method calculates the quantity exactly but eats up time and computer resources in the process. There is another method that is less computationally expensive, but it yields only an approximation of the binding free energy.
Zhang and Ding’s DeepBAR approach traditional chemistry calculations with recent advances in machine learning to compute binding free energy exactly — but with a fraction of the calculations needed with previous methods.
In using deep generative models, the researchers were borrowing from the field of computer vision. “It’s basically the same model that people use to do computer image synthesis,” Zhang said. “We’re sort of treating each molecular structure as an image, which the model can learn. So, this project is building on the effort of the machine learning community.”
Adapting a computer vision approach to chemistry was DeepBAR’s key innovation, but the crossover also raised challenges. “These models were originally developed for 2D images,” Ding said. “But here we have proteins and molecules — it’s really a 3D structure. So, adapting those methods in our case was the biggest technical challenge we had to overcome.”
Filed Under: Drug Discovery, Drug Discovery and Development, RD



Tell Us What You Think!
You must be logged in to post a comment.